꽤 오랫만에 쓰는 시리즈물의 최신 에피소드가 되겠음. 지금까지의 ‘계명’ 은..
1. 단백질은 단백질마다 다 다르니 그들의 특성을 잘 파악할지어다.
2. 폴리펩타이드 결합은 언젠가 끊어질지니 단백질 분해효소에 유의할지어다
4.친화크로마토그래피와 친하게 지내라 이었다.
그놈의 다섯번째 계명은 무엇인지 궁금해하시는 분이 많았다. 제 5계명은 다음과 같다.
이온교환크로마토그래피를 사랑하라
친화크로마토그래피를 했습니다. 그 다음에는요?
앞에서 말했듯이 요즘의 단백질 정제는 단백질에 달아놓은 태그를 특이적으로 인식하는 레진을 이용한 친화크로마토그래피로 시작하는 것이 보통이다. 때로는 친화크로마토그래피 한번으로 (자신이 사용하기에) 충분히 높은 순도의 단백질을 얻는 경우도 많다. 특히 단백질 발현이 잘될 경우라면 더더욱 그렇다. 그러나 세상일이 언제나 그렇게 쉽게 풀리지만은 않는 법이므로 친화크로마토그래피 한번으로는 충분히 정제된 단백질을 얻지 못하는 경우가 많다. 그럴 때는 어떻게 해야 하나.
이럴때는 기존의 단백질 정제에서 기본이 되는 두가지의 서로 상이한 원리의 크로마토그래피가 등장할 차례이다. 이번에 다룰 것은 이온교환크로마토그래피 (ion-exchange chromatography)
뜨거운 단백질과 쿨시크한 단백질
이제쯤이면 귀에 못이 박히듯이 들어서 지겨워질 이야기가 있다. 뭐냐고? “단백질은 단백질마다 그때그때 달라요” 다. 즉 단백질은 단백질마다 크기도, 모양도, 어떤 물질에 붙냐도, 그리고 표면의 극성도 다 틀리므로 모든 단백질을 동일한 방법으로 정제할 수는 없다. 즉, 단백질 정제라는 것은 근본적으로 우리가 목적하는 단백질의 고유한 성질을 이용하여 다른 단백질과 구분해내는 방법이다. 그런 다른 성질 중에서 이용할 수 있는 것은 ‘어떤 물질에 붙냐’ (이것을 이용하는 것이 바로 지난번에 알아보았던 ‘친화크로마토그래피’이다), 혹은 ‘표면의 극성’ 이나 ‘단백질의 크기’ 등등이다. 단백질의 크기에 대해서는 오늘 이야기하지 않을것이고, 단백질의 특성으로 중요한 것은 ‘표면의 극성’ 이다. 단백질은 근본적으로 아미노산이 서로 이어져 있는 폴리펩타이드이고, 각각의 아미노산은 종류에 따라서 서로 다른 성질을 가진다.

즉 중성 pH (7.0) 을 기준으로 할때 Lys, Arg 류는 양성을 띄고 있고 Asp, Glu 등은 음성을 띄고 있음. Ser, Thr 등은 그냥 극성만 띄고 있을뿐. Leu, Tyr, Phe, Trp 등은 비극성. 그런데 막단백질을 제외한 상당수의 단백질은 일단 수용액상에 녹아 있고, 자연스럽게 표면으로 노출되는 아미노산은 양성, 음성으로 차징되어 있는 넘, 혹은 일부 극성 아미노산. 물론 비극성 아미노산들도 일부 노출되는 것들이 있긴 하나 어쨌든 수용액상에 녹아있는 아미노산의 표면에는 이렇게 양성 혹은 음성 전하를 띄고 있는 넘들이 상당수를 이루게 되고, 이것의 조성에 따라서 단백질전체가 양성 혹은 음성을 띄고 있을 수 있으며, 이것은 단백질마다 다 각각 틀려요라는 이야기. 그런지 안그런지 제가 직접 보여드리겠습니다 그만해 ㅇㅇㄷ의 에너지는 이미 0이야! 
뭔 단백질인지 모르겠지만 단백질이 몽땅 시퍼래..여기서 시퍼런 색은 표면에 양성극성을 띈 아미노산이 많다는 이야기고, 흰색은 비극성, 빨간색은 음성극성이 많다는 이야기이다. 그런데 뭔 단백질인데 이렇게 표면에 양성을 많이 띄냐고.
.\ 
히스톤…DNA에는 강력한 – 극성을 띈 인산기가 있으니 여기에 붙으려면 천상 + 이 되야죠 뭐…
반면 아래와 같이 전반적으로 표면에 – 극성을 띈 아미노산이 많은 단백질, 즉 전반적으로 마이너스 극성을 띈 단백질도 있다.

그래서 ‘단백질끼리 서로 극성이 다양하게 틀린’ 성질을 이용하여 서로 다른 단백질을 분리해보자는 것이 이온교환크로마토그래피의 기본 원리가 되겠다.
그러나 앞에서 말한 친화크로마토그래피의 경우 특정한 태그를 달았을 경우에는 단백질의 종류와는 대개 상관없이 균일한 프로토콜로 정제가 가능하지만, 이온교환크로마토그래피부터는 그런 것은 통하지 않음. 즉, 단백질이 강하게 + 를 띄는 것도 있고, 대충 엉성하게 + 를 띄는 것도 있고, 한쪽은 +를, 반대쪽은 – 를 띄는 것도 있고, 엉성하게 – 를 띄는 것도, 강하게 – 를 띄는 것도 있는데 이를 어떻게 하나의 프로토콜로 정제한다는 것인가?
즉, 단백질마다 다 그 정제 조건은 틀리므로 이제부터는 자기의 완소단백질에 맞는 고유한 정제조건을 확립하는 것을 배워야 한다고. 일단 필요성은 이만하면 됐고, 이제부터 ‘이온교환’ 이라는 알쏭달쏭한 단어에 대해서 좀 알아보도록 하자.
양이온 교환과 음이온 교환
이온을 교환한다 (ion exchange) 라는 말이 잘 와닫지 않는 쪼랩연구자가 있을지 몰라서 부연설명하도록 하는데, 간단하게 생각하면 양이온은 음이온에 붙고, 음이온은 양이온에 붙는다 내지는 산은 산이요 물은 물이냐 수준의 이야기를 다르게 표현한 말에 지나지 않는다.

(c) 김성모 이런 표정을 짓고 계시는 분들도 있을듯 싶어서 부연 설명을 하자면
음이온 교환(anion exchange) : 음성 극성을 띄고 있는 이온이 양성 극성을 띈 레진에 붙는다. 즉 붙는 단백질은 : 전체적으로 음성을 띄고 있으며, 음성을 띈 정도가 높을수록 강하게 붙는다. 음이온 교환 크로마토그래피에 사용되는 주된 레진인 DEAE나 Quatenary Anime은 아래 화학식에서 느껴지듯이 강한 ‘양성’을 띄고있다. 따라서 여기에 붙는 단백질은 ‘음성’ 이 되겠다 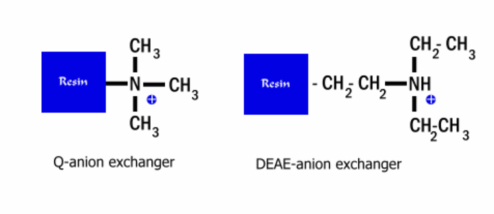
양이온 교환 (cation exchange) 양성 극성을 띄고 있는 이온이 음성 극성을 띈 레진에 붙는다. 즉 붙는 단백질은 : 전체적으로 양성을 띄고 있으며, 양성을 띈 정도가 높을수록 강하게 붙는다. 
그렇다면 ‘교환’ 이란 무슨 이야기인가? 결국 우리가 이런 것에 관심을 두고 있는 이유는 ‘단백질을 순수정제’ 하기 위함인데, 단백질을 이런데 떡 붙여놓고만 있으면 안된다. 즉 회수를 해야지. 그렇다면 회수를 하려면 어떻게 해야하나? 여기서 ‘교환’ 의 의미가 등장하게 되는데, ion exchange에 붙어있는 단백질 등을 떼기 위해서는 이보다 더 강력한 ‘뭔가’ 를 이용해서 떼내야 한다. 만약 ‘음성을 띄고 있는 단백질이 붙어있는 음이온 교환수지’ 이라면 음이온, 가령 Cl(-) 이온을 이용하여 기존에 붙어있는 단백질을 이보다 더 극성을 가지는 이온으로 ‘교환’ 될 수 있다.
이때 음이온 교환수지에서 목적하는 단백질을 떼네는데 사용하는 이온을 카운터이온 (Counter-ion)이라고 불리고, 가령 소금물 (NaCl) 용액에서 음이온 교환수지에서는 염소이온이 카운터이온이 된다. 양이온 교환수지라면? 양이온, 예컨대 Na(+) 이온을 가하면 + 극성을 띈 단백질이 Na(+)과 ‘교환’ 되고 단백질은 떨어지게 된다. 그리고 카운터이온은 소디움 이온이 되는거고.
즉, 앞에서 단백질들은 가령 중성 (pH=7.0) 에서 매우 강한 음성부터 매우 강한 양성을 띄는 넘들이 있을 것이고, 매우 강한 음성을 띄는 넘들은 음이온 교환수지에 ‘강하게’ 붙을 것이고, 거의 중성에 가깝지만 약간 음성을 띄는 넘들은 ‘슬쩍’, 그리고 양성을 띄는 넘들은 아예 붙지 않을 것이다. 이런 상태에서 점점 염 농도, 예컨데 Cl(-) 농도를 높인다면? 처음에는 ‘슬쩍’ 붙어있는 넘들이 ‘버..버틸 수 없다!’ 를 외치며 떨어질 것이고, 점점 염 농도를 높일수록 강하게 붙은 넘들이 떨어지기 시작한다. 이렇게 염 농도를 높이면서 나오는 것들을 별도의 튜브에 받으면 단백질을 극성에 따라서 분리할 수 있다. 
참 쉽죠? 왠지 이 아저씨의 얼굴이 떠오르면 지는거다
일단 소금기부터 빼고 오세요 고갱님
따라서 이온교환크로마토그래피는 보통 염농도가 매우 낮거나 거의 없는 상태로부터 샘플을 이온교환수지가 들어있는 컬럼에 붙이고 이를 점점 염농도를 높여가면서 나오는 국물(..)을 받는 것이라고 정의하면 되겠다. 그 이야기인 즉슨, 일단 이온교환크로마토그래피를 하기 위해서는 염 농도가 낮은 수준의 버퍼에 들어있어야 하겠다. 가령 1M 정도의 NaCl 이 들어있는 버퍼에 단백질이 들어있다면 대개의 단백질은 이온교환수지에 붙어볼 겨를도 없이 죽 빠져버린다. 가령 앞에서 설명한 친화크로마토그래피에 흔히 사용하는 Ni-NTA 혹은 GST 등과 같은데서 사용되는 버퍼는 대개 500mM 이상의 높은 염농도를 자랑하는데, 일단 친화크로마토그래피를 걸고 나서 SDS-PAGE 를 걸어보니까 잡스러운 단백질들이 많이 있어서 이걸 좀 더 정제해보겠어요~ 라고 그대로 그걸 이온교환크로마토그래피에 걸어버리면~ 아마도 대개의 경우 님의 목적단백질과 잡 단백질은 바로 컬럼에 붙지 않고 빠져버릴 것이다. 그러므로 이온교환크로마토그래피를 위해서는 일단 단백질이 들어있는 용액내의 염 농도를 낮추어야 한다.
(중요하니까 볼드치고, 밑줄도 쫙하고 빨간색으로 처리했다. 이러면 그만큼 중요한 줄 알아라. 대개의 이온교환크로마토그래피가 망했어요 하는 양반들은 이게 잘 안되서이다.)
그렇다면 소금기를 빼는 방법에는 어떤 방법이 있는가? 가장 일반적인 방법이라면 투석 (Dialysis)가 있겠고, 
만약 샘플의 부피가 그닥 많지 않다면 젤여과 크로마토그래피의 원리를 이용한 각종 탈염칼럼 (Desalting Column)을 쓸 수도 있다.

각각에 대해서 자세한 설명을 구구절절히 하는 것은 좀 그렇고, 몇가지 주의할 점을 들어보면
– 투석을 할 때는 버퍼와 샘플의 비를 생각하는게 좋은데, 가령 100ml의 버퍼가 들어있는데에 10ml 의 투석을 한다면, 10ml 안에 있는 고염용액이 아무리 잘 투석되어 봐야 10% 로밖에 줄어들지 못한다. 따라서 가능한 버퍼와 샘플의 부피비를 늘리는 것이 좋다. (그런데 그렇다면 버퍼를 많이 만들어야 되잖아..네 맞구요 고갱님, 단백질 정제계에 뛰어든 순간 당신은 하루하루 버퍼만드는 기계일 뿐이죠 후후후)
– “투석은 오래 걸리잖아요 히잉~ 근데 내 단백질은 워낙 불안정해서 오래 못버텨요. 어떻게 투석을 빨리 하는 방법이 없을까요?” 버퍼를 자주 교체하세요 고갱님.
컬럼에 로딩
그래서 이제 샘플이 준비되었다면 컬럼에 로딩할 차례! 이온교환크로마토그래피는 직접 레진을 패킹하는 오픈컬럼으로 수행할 수도 있으나, 요즘은 대개 FPLC시스템과 연계되어 사용되는 고성능-고해상도의 컬럼들이 존재한다. 가장 대표적인 것이라면 Pharmacia 사의 제품이었으나 여러단계의 기업인수합병을 거쳐서 이제는 GE에서 나오는 ‘Mono-Q/Mono-S’ 시리즈를 들 수 있다.

이온교환크로마토그래피와 아마 다음차례에 소개할 젤 여과 크로마토그래피 (Gel Filtration Chromatography) 와의 결정적인 차이라면 샘플을 로딩하는 방식에 있을텐데, 젤 여과 크로마토그래피에서는 샘플을 농축하여 최소한도의 부피로 샘플을 로딩하는 것이 필수지만 이온교환크로마토그래피는 그딴거 필요없다. 즉 샘플의 부피는 얼마가 되든, 해당 컬럼에 우리가 정제하려고 하는 단백질이 붙을 수가 있다면 로딩이 가능. 따라서 이온교환크로마토그래피에서는 농도가 매우 낮은 샘플의 경우에도 특별히 농축이 필요없이 로딩한 후, 샘플을 농축하는 효과도 같이 얻을 수 있다. 따라서 수십 ml 로부터 심지어는 수백 ml – 리터수준의 샘플을 로딩이 가능한데, FPLC등의 시스템을 이용할때는 다음과 같은 소위 ‘SuperLoop’ 라는 것을 이용하기도 한다. 
그리고 앞에서 이온교환크로마토그래피는 비교적 초기 단계에서 이용하기 편하다고 했지만, Mono-Q 등과 같은 고가의 FPLC용 컬럼의 경우 일차 정제단계 (친화크로마토그래피) 등을 거친 다음의 후속 정제에 사용하는 편이 낫다. 가끔 세포를 파쇄한 ‘국물’을 센돌이 돌려서 바로 Mono-Q 등에 로딩하는 경우도 보곤 하는데, 이 경우 고가의 컬럼을 망가뜨릴 염려가 있으니 주의. 이런 ‘국물’ 에는 단백질 뿐만 아니라 엄청난 양의 DNA 등이 포함되어 있는데, DNA는 Mono-Q 에 매우 잘 붙고, 1M 정도의 대개의 단백질이 다 떨어질 높은 염농도에도 떨어지지 않는다. 따라서 이런 짓거리 한두번 하다보면 컬럼이 망가지기 쉽상이니 이런짓 하지말자. -.-;;;
워싱을 워시워시
일단 컬럼에 우리가 원하는 단백질이 들어있는 ‘국물’ 을 한방울도 남기지 않고 흘려넣으면 결국 다음 상태인데,
– 컬럼에 붙지 않는 대개의 단백질은 붙지 않고 떨어져나가고
– 원하는 단백질과 극성 성향이 유사한 단백질은 붙어있는 상태, 여기서 이온농도를 높여서 원하는 단백질들을 떨어뜨리기 전에, 일단 컬럼에 붙지 않는 대개의 단백질을 ‘학실’ 하게 제거할 필요가 있다. 즉, 컬럼에 로딩한 단백질이 녹아져 있는 버퍼, 혹은 이보다 조금 더 이온농도가 높은 버퍼를 이용하여 컬럼에 흘려준다. ‘얼마나?’ 라고 말하자면 흔히 컬럼의 부피(CV)의 몇 배라고 이야기하는 메뉴얼이 많지만, 여기서는 그딴 것 없고 ‘더이상 단백질이 흘러나오지 않을 정도’ 로 주구장창 워싱을 해준다. UV Monitor로 단백질 농도를 추적할 수 있는 FPLC 등을 이용한다면 쉽게 가늠을 할 수 있겠고, 그렇지 않다면 컬럼을 거쳐 나오는 Fraction에 단백질 정량에 사용하는 Bradford Solution을 조금 넣어서 색깔이 퍼렇게 변하냐 안하냐로 알아볼수도 있겠다. 여튼, 더이상 단백질이 나오지 않을 때까지 확실하고 철저하게 워싱을 해주는 것은 기본 되겠다. 특히 단백질의 로딩량이 많을 경우에 이런 문제가 더할 수 있으므로..
아무튼 워싱은 확실하고 철저하게.. 물론 당연한 이야기지만 워싱하는 조건에서 원하는 단백질이 조금이라도 흘러나오면 -.-;; 안된다.
자, 이제 우리가 레진에서 헤어져야 할 시간
이제 다음 스탭은 우리가 목적하는 단백질을 카운터이온 농도를 올려서 떼어낼 차례이다. 여기서 알아두어야 할 것은 컬럼에 아예 붙지 않는 단백질을 제거한 이후에도 여전히 수많은 단백질들이 컬럼에 붙어있고, 이들을 미세한 극성의 차이에 의해서 분리하기 위해서는 카운터이온 농도를 어떻게 올리냐가 문제가 된다는 것이다. 가령 우리가 원하는 단백질이 pH 7.0 의 버퍼에서 약 200mM 정도의 NaCl 이 들어있을때 음이온 교환수지에서 떨어진다고 하자. 이를 분리하기 위해서는 어떻게 해야할까? 가령 1M 의 NaCl을 바로 흘려버리면 200mM 정도의 NaCl에서 떨어지기 시작하는 우리 단백질도 당연히 나오겠지? 그러나 그 경우에는 100mM, 150mM 등 우리의 단백질보다 극성이 약해서 낮은 염농도에서 떨어지는 단백질과, 300mM, 400mM, 500mM…등 우리보다 세게 붙은 단백질도 동시에 레진에서 떨어져 버릴 것이고, 결국 이러한 단백질들간의 분리가 이루어지지 않는다.
그렇다면 우리가 원하는 단백질이 어디서 떨어지는지를 알아야 되는데, 미지의 단백질이라면 어떻게 그걸 아냐고..그래서 사람들이 생각해 낸 것이라면 두 개의 펌프를 이용하여 서서히 염농도를 올려가면서 단백질을 점점 높은 염 농도에서 우려내는 것. 소위 말하면 농도구배 (Gradient)를 주는 것이다. 그렇다면 어떻게 농도구배를 주는가? 옛날에는 이런 기구를 사용하였다.


즉, 동일 조성인데 단지 염 농도만 다른 두 가지 버퍼를 만들고 (염 농도가 0인 것을 버퍼 A라고 부르고, 염 농도가 1M 인 것을 버퍼 B라고 부르자) 요렇게 생긴 용기에 넣는다. 두 용기는 튜브로 연결되어 있고, 컬럼으로는 낮은 염농도의 버퍼가 연결되어 있다. 이렇게 해서 버퍼를 계속 흘려주고, 낮은 농도의 버퍼에서 계속 섞어주면, 염 농도는 처음 낮은 농도부터 시작하여 높은 농도로 계속적으로 증가되게 된다. 믿을랑가 모르겠지만 생화학의 황금기 시절에는 다들 이렇게 단백질을 정제하였다. 그러나 요즘은 대개 이런 시스템보다는 직접 두 개의 버퍼를 펌프에 연결하고, 이들을 컴퓨터 프로그램에 의해서 직접 컨트롤하는 시스템을 사용한다. 
즉, (1)번에 ‘버퍼 A’ 과 ‘버퍼 B’ 를 별도로 연결하면 컴퓨터에 연결된 컨트롤러인 ‘3’ 에 의해서 적절한 배합비에 따라서 적절한 농도구배를 만들어준다. 여튼, 결과적으로 우리가 얻는 것은.. 
여기서 파란색은 단백질 (A280) 을 모니터링한 것, 적색은 염농도를 모니터링한 것이다. 제일 먼저 나오는 피크는 해당하는 컬럼에 붙지 않는 것, 그리고 낮은 염 농도에서 떨어지는 넘들은 해당 컬럼에 약하게 붙는 것이고, 뒤로 갈수록 해당 컬럼에 강하게 붙는 것이다. 즉, 이렇게 Gradient를 걸면서 샘플들을 일정한 부피로 분별을 하는데, 물론 이 작업은 대학원생 학부생;;; 을 시킬수도 있겠으나 (-.-;;) 이런 기구가 있으니 이것이 바로 Fraction Collector이다.

이게 없었으면 얼마나 많은 대학원생이 콜드룸에서 날밤새다 감기에 걸렸을까 생각해보자
그래서 결국 이온교환크로마토그래피를 끝난 후에 얻는 결과물은 위와 같은 크로마토그램 (FPLC 등의 시스템을 사용했을 경우) 및 여러개의 단백질이 담겨있는 튜브일 것이다. 그렇다면 여기서 우리가 원하는 단백질은 어디에 담겨있는가?
Assay
사실 요즘에는 일단 단백질을 자연계에 존재하는 양의 수십, 수백, 수천배로 오버익스프레션을 하고, 그리고 이온교환크로마토그래피를 하기 때문에, 그리고 대개 이미 알려진 유전자에 대해서 단백질을 발현하여 단백질 정제를 하기 때문에 우리가 원하는 단백질이 SDS-PAGE를 걸면 어떤 건지 대충은 아는 상태에서 정제를 하는 편이다. 그러나 이것은 이러한 것들이 보편화된 다음의 이야기이며, 자연계에 존재하는 단백질을 세포를 수십, 수백리터 배양해서 정제하던 시절에서는 전혀 이런 것을 기대할 수 없었다. 심지어 생화학적으로 알려진 수많은 단백질은 SDS-PAGE라는 실험기술이 존재하기도 전에 정제되었다!! ‘ㄷㄷㄷ 그러면 어떻게 단백질을 정제함?’ 이라고 생각할런지 모르겠지만, 그 시절에서 수많은 프랙션 중에서 우리가 원하는 단백질이 들어있는 것을 알아내기 위한 유일한 수단은, 해당 단백질의 활성측정, 즉 assay였다. 가령 우리가 원하는 단백질이 무슨 기질 A를 B로 변환시키는 효소라고 할때, 결국 하는 일은 기질 A와 수많은 Fraction을 가지고 효소반응을 실시한 후 B의 양을 정량하여, 어떤 fraction에 원하는 활성을 가진 단백질이 가장 많이 존재하는지를 알아내는 것만으로 원하는 Fraction을 추정해 나간다는 이야기이다.

바뜨, 요즘은 대개의 경우 SDS-PAGE로 원하는 목적단백질의 밴드를 정제 전에도 확인한 상태에서 이온교환크로마토그래피를 걸기 때문에, 웬만한 경우에는 Fraction에 대해서 모두 효소활성을 측정할 필요가 없이 그냥 SDS-PAGE만으로 원하는 단백질이 들어있는 Fraction을 알 수 있는 경우가 허다하다! 
요즘은 대개 이런식으로 SDS-PAGE만으로 원하는 단백질과 유사한 사이즈의 단백질을 가지고 추정을 한다. 그러나 조심해야 할 것이 이러다가 원하는 단백질과 사이즈가 비슷한 단백질을 잘못 정제하지 않았는지 확인을 해봐야 한다는 것! (주변에서 세포를 파쇄하는 것을 돕기 위해 넣어준 Lysozyme을 목적단백질인줄 알고 잘못 정제하여 Lysozyme 결정을 싱크로트론에 들고간 사람을 본 적이 있다 ㄷㄷㄷ) 효소라면 활성 assay, 마땅한 활성 assay가 없는 경우라면 Mass Spec 등으로 우리가 정제하고 있는 단백질이 과연 목적하는 단백질인지를 꼭 확인할지어다.
‘그래서 내 단백질을 정제하는데 음이온 교환을 써야 하나염 아니면 양이온 교환을 써야 하나염?’
즉 결국은 우리가 원하는 단백질이 붙는 교환수지를 사용하여 단백질을 정제하는 것이 기본인데 (물론 역으로 ‘우리가 원하는 단백질이 붙지 않는’ 교환수지를 이용하여 우리가 원하는 단백질 외의 상당수의 단백질을 제거할수도 있다. 그러나 뭐 일단은 ‘붙이는 것’ 자체가 기본이다) 과연 우리가 원하는 단백질이 현재 녹아있는 버퍼 조건에서 어떤 이온교환 수지에 붙을지를 어떻게 알 수 있을까? 걍 실험해서? 물론 그게 정답이긴 하지만 -.-;; 그래도 우리는 요즘 대개 클로닝되고 과발현된 단백질을 이용하여 단백질을 정제하고 있기 때문에, 우리가 원하는 단백질의 단백질 조성을 알고 있는 경우가 대부분이다. 여기서 포인트는 우리가 정제하고 있는 단백질의 아미노산 조성을 이용하여 ‘대략적인’ 단백질의 isoelectric point, 즉 net charge가 0 이 되는 pH를 알 수 있다. 일단 이 링크로 ㄱㄱ 
웹사이트의 텍스트박스에 정제하고 있는 단백질의 아미노산 서열을 붙여넣고 ‘Compute Parameters’를 누르면.. 
여기서 눈여겨볼게 ‘Theoretical pI’ 인데 이 단백질의 경우에는 5.29 이다. 이 이야기는 pH가 5.29일때 이 단백질의 net charge는 0이라는 것이다. 이것보다 pH가 올라가면, 전체 단백질의 charge는 마이너스로 가고, 이것보다 pH가 낮아지면 전체 단백질의 charge는 + 쪽으로 움직인다.
생화학이나 일반화학시간에 졸면 이렇게 된다
이런 분들을 위해서 생화학책 혹은 일반화학책에 나오는 아미노산 적정그림 다시 첨부해봤다. 
여튼 위의 예에서는, pI가 5.29이므로 만약 이 단백질이 중성인 pH 7.0 근처의 버퍼에 녹아있는 경우에는 당연히 (-) charge를 띨 것이며, 이 단백질은 아마 어디에 붙을까요? 정답은 음이온 교환수지.
만약 다른 단백질을 가지고 pI를 계산했을때 약 8-9 정도가 나왔고, 역시 동일한 pH 7.0 근처의 버퍼에 있을때는 (+) charge를 띌 가능성이 높으므로, 이 단백질은 아마도 양이온 교환수지에 붙을 가능성이 높을 것이다. 그러나, 이러한 방법으로 ‘이론적인’ pI를 예측하는 것은 어디까지나 전체 아미노산의 조성을 가지고 계산하는 예측이며, 이것만으로 단백질의 실제상황에서의 net charge, 그리고 해당 단백질이 양이온 교환수지 혹은 음이온 교환수지에 붙을지 안 붙을지를 정확하게 알 수는 없다. 왜냐하면 단백질의 실제 상황에서의 net charge에 기여하는 단백질은 표면에 노출되어 있는 단백질인데, 해당 상황에서는 그런 것은 신경안쓰고, 그냥 모든 단백질의 조성으로 어림잡았기 때문이다. 그리고 우리가 원하는 단백질이 실제로 다른 단백질과 혼합되어 있을때 다른 단백질혹은 다른 물질과 결합되어 있거나 (가령 DNA 결합 단백질이라면 DNA와 결합되어 있는 경우라든지) 하는 경우에는 계산된 이론 pI와는 다르게 작용할 가능성도 있다.
따라서 이론적인 계산은 어디까지나 이론적인 계산으로 생각하고, 다르게 나올 가능성이 있다는 것을 염두에 두고 고려하는 것이 좋다고. (실험하라고요)
버퍼의 pH를 잊지마!
위에서 본 것처럼 특정한 단백질의 net charge가 어떻게 될지는 현재 그 단백질이 녹아있는 버퍼의 pH에 결정적으로 좌우된다. 즉, 동일한 단백질도, pH에 따라서 양이온 교환수지에 잘 붙는 단백질이 될 수도 있고, 음이온 교환수지에 잘 붙는 단백질이 될 수도 있다. 또한 같은 음(양)이온 교환수지에 붙어도, pH에 따라서 100mM의 염농도에서 떨어져 나올수도 있지만 200-300mM에서 떨어져 나올 수도 있다. 그러므로 앞에서 알아본 이론적인 pI의 경우에는 단순히 단백질이 어떤 이온교환수지에 붙을 것인가를 예측하는 것에서 더 나아가서 이를 좀 더 ‘잘 붙이려면’ 어떻게 해야하는지에 대해서도 알 수 있다.
가령 우리가 이론적으로 계산된 pI가 약 6.5 인 단백질이 있다고 하자. 우리는 현재 버퍼로 pH 7.0 인 것을 사용하고 있는데, 음이온 교환수지에 단백질을 붙여보니, 거의 이 단백질이 붙지 않았다. 이 경우에는 어떻게 해야하나?
만약 단백질의 이론적인 pI 계산이 실제와 유사하다고 가정하면, 해당 단백질은 pH 7.0 에서 ‘아주 약간만’ 네거티브한 극성을 띄고 있을 것이다. 그런 경우에 단백질의 극성을 높이려면? 아마 버퍼의 pH 를 좀 더 높여주면, 이 단백질은 해당 버퍼에서 좀 더 net charge가 낮아질 것이고 음이온 교환수지에 좀 더 잘 붙을 가능성이 농후하다. (물론 역시 해보기 전에는 100% 확신은 못한다 ㅋ)
반대로, pH를 pI보다 좀 더 낮추어 주면, 이제 해당하는 단백질은 net charge가 +이 되고, 이번에는 양이온 교환수지에 잘 붙을수도 있다. 아무튼 이온교환수지를 통해서 단백질 정제를 최적화하기 위해서는 버퍼의 최적 pH를 찾는 것이 중요하며, 이것을 정복하는자, 이온교환수지를 정복하는 것임을 잊지 말지어다. 물론 단백질은 ‘다 그때그때 달라요’ 를 잊지 말고..
요즘은 단백질 정제를 아무나 한다고?




(c) 엉덩국
요즘 단백질 과발현과 간편한 친화크로마토그래피의 보급에 따라서 단백질의 정제는 아무나 할 수 있는 것으로 흔히 오인되곤 한다.그러나, 구조생물학이나 생물물리학 연구에 필요한 순도높은 단백질의 정제, 혹은 단백질 복합체의 정제에는 여전히 이온교환크로마토그래피나 젤여과 크로마토그래피등과 같은 기존에 확립된 테크닉이 필요하며, 이러한 ‘고전적’ 인 단백질 정제기술에 얼마나 능하느냐가 당신을 단백질 정제기술의 전문가인지, 아니면 그저 간단한 단백질 정제하는 시늉만 내 본 사람인지를 구별하는 기준이 될 수 있다. 하드코어 단백질 전문가라면 당연히 이런 고전적인 단백질 정제기술에 누구보다도 자신감을 가지고 있어야 한다는게 내 생각. 자 우리 모두 하드코어가 됩시다
P.S. 전재를 하시는 분은 옮기는 상관없으나 저작권에 문제가 될 수 있는 패러디 부분은 자제해주시길 ㄷㄷ

안녕하세요. 몇가지 질문이 있는데요.
1) 단백질이 multimer을 form할 경우에 ion-exchange column을 썼을때 pure한 monomer이랑 같이 elution이 되나요? 아니면 나중에 다른 salt gradient 에 따라 elution이 되나요?
2) MES를 buffer로 쓸 경우 conductivity에 영향을 끼치는거 같은데.. 혹시 왜그런지 설명해줄 수 있으신가요? 감사합니다!
(1)역시 이런건 단백질에 따라 틀리지만, 만약 multimer가 안정되게 유지되고, 호모지너스하면 ion exchange로 분리하긴 힘들죠. 이 경우에는 gfc가 답
2) mes 의 concentration에 따라 틀린데 뭐 100mM 이렇게 높으면 당연 conductivity에 영향끼침. 버퍼는 버퍼링이 될 최소농도로..
감사합니다!
천재이신가요? 진짜 쉽고 재밌게 설명해놓으셨네요.
아이노산 107개인 단백질 정제하는데,아미노산 700개 단백질 보다 더 수월할까요?
‘단백질은 그때그때 달라요’ 이므로 일률적으로 말하기 힘듭니다. 만약 107개짜리 단백질이 안정성이 떨어져서 미친듯이 분해되는 단백질이라면…ㅎㄷㄷ
Kir3.1 cytoplasmic domain입니다. Cardiomyocyte에서의 양도 적고, signal이 오면 발현되는 protein입니다.지금 E-coli expression중인데 걱정입니다. 작년 10월에 Kir3.1 whole length를 했는데,inclusion body (insoluble) 상태로 되었 거든요.그래서 필요한 부분만 다시 expression 하고 있읍니다만…답변 정말 감사합니다.
아 이온채널 ㄷㄷㄷ
Full length는 아마 대장균에서 발현한다는 것 자체가 무리일 것으로 생각되구요, Cytoplasmic domain 정도는 예상되는 cytoplasmic domain간의 interaction 을 잘 고려하여 construct를 만들면 될 수 있을지도 모릅니다만…
cytoplasmic domain은 G-loop으로 연결되어 multimer를 구성하고 있읍니다. 선생님 말씀을 들으니 그래도 좀 안심이 됩니다. 감사합니다.
특허소송 대리 중인데 배경기술 이해하려고 찾아 들어왔습니다. 정말 도움 많이 되었습니다. 감사합니다.
다음 계명은 언제쯤 출간될까요.. 빨리 읽고싶어지네요
안녕하십니까. 효소도 단백질이기 때문에 똑같은 과정으로 정제하면 될까요? 교수님께서 처음 과정으로 sepadex로 분자량 별로 분리먼저 하라고 하시는데..솔직히 단백질 하는 실험실이 아닌데 갑자기 효소 정제를 하라고 하셔서 너무 힘듭니다. tannase를 분리하고 있는데요, 버퍼로 균질화하여 하루간 버퍼 추출 후 ammonium sulfate 30~80%로 cut했고, 투석하여 잔여 염을 제거하였습니다. 이것을 겔 크로마토그램으로 가야될지, sephadex로 가야될지…..도와주세요 … 실험실에 혼자뿐이라 힘드네요…
이 시리즈를 잘 읽으셨으면 감이 잡힐텐데, 젤 여과 크로마토그래피는 뒤에 하는 것이 정석입니다. 그리고 ‘sephadex’ 라고 하시는것이 정확히 sephadex 라는 레진을 사용하는 젤 여과 크로마토그래피인지 DEAE-sephadex 와 같은 음이온 크로마토그래피인지 확실하지 않은데, 아래 논문을 보면 DEAE 같은 음이온 교환 크로마토그래피겠네요.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3848197/
일단 문헌 (종이 틀려도 대략적으로 단백질의 특성은 유사한 경우가 많아요) 에 나온대로 투석후 DEAE-sepharse..아니 다른 anion exchanger 가 있으면 굳이 DEAE 안써도 됩니다. Q-Sepharose, 혹은 Mono-Q 등을 써도 됩니다. 여튼 anion exchanger 를 이용하여 음이온 교환크로마토그래피 하세요.
감사합니다^^ 교수님은 반대로 또 하라시네요 ㅜ 시키는대로 해야죠 뭐 흔한 대학원생이니…ㅋㅋㅜㅜ 아무튼 글 읽고 이론적으로도 많은 도움 얻어갑니다^^
안녕하세요. 써주신 글 덕분에 잘 이해할 수 있었습니다. 순수 분석화학을 전공하고 NMR기기 분석만 하다가 처음으로 단백질정제라는 것을 하게되어 많이 헤매고 있었는데 많은 도움이 되었습니다.
질문하나만 드릴께요.
단백질이 하나 있습니다. pI는 3.68이구요. IEX를 하려고 합니다.
제가 이해하기로는 위 내용상으로는 Q나 DEAE 같은 anion column을 사용하는것이 맞을까요? 실험에 사용할 컬럼선택부터 Buffer조성까지 알아서
디자인을 해오라고 하는데 탈모가 생길 지경입니다.
안녕하세요 효소공부하고 있는 학생입니다. 효소를 정제 후에 바로 aliquot하고 급속동결 해 놓지 않고 다량으로 두세번 얼렸다 녹였다를 반복했는데요. 이 과정에서 효소의 활성이 거의 없어질 수도 있는것인가요?
반복적인 얼렸다 녹였다는 효소 활성에 매우 안좋습니다. 제한효소나 pcr 효소 등이 50% glycerol이 들어있는 버퍼에 들어있는것도 이런 이유때문이죠.
고농도의 염에서 올라가는 peak은 어떠한 걸로 예상할 수 있을까요?
핵산같은 것인가요?
Anion이면 핵산일 가능성이 높죠..1M 이상에서 떨어집니다
감사합니다..ㅎㅎ
질문하나 하겠습니다.
Ni-NTA Column 후 SDS PAGE를 통해 확인할때요 washing 하기 전의 샘플의 밴드는 여러개가 보이는데 washing 후 샘플에는 밴드가 구분되기 힘들게 잡밴드들이 끌림현상으로 나타나고 elution 샘플에서는 아무 밴드도 나타나지 않았는데요, 이론대로라면 washing후 밴드와 elution 밴드가 어떤 형태로 나타나야하나요?
https://www.thermofisher.com/order/catalog/product/25214
재밌게 설명해주셨네요ㅕ 생화학시간에 졸지 않았는데 왜이리 어려운지
포스팅 꼼꼼히 읽어보았습니다. 좋은 글 감사드리며 질문 몇가지 드립니다.
지금 제가 가지고 있는 protein의 PI가 공교롭게도.. 딱 7.0입니다 (물론 protparam 이론상값입니다). 지금 저의 애매한 상황에서, 확실한 이온컬럼에 binding시키기 위해서는 buffer의 pH를 올리거나 내려서 protein의 PI값을 조정하면 되는 건 위에 포스팅해준 글에서 잘 보았습니다.
그런데 궁금한 점이, 만약에 buffer의 높고 낮은 pH로 인해 제 protein의 denaturation(변성)의 우려가 됩니다. (protein은 pH,온도 등이 민감하니까요..)
두번째 질문입니다.
이건 글을 읽고 이해가 안가는 부분인데요-
Ni-NTA column을 걸고 난 후 2차 정제 전에, 탈염과정을 거치는 이유가 protein들이
column을 다 통과해버리기 때문이라고 해주셨는데, 이 부분이 이해가 가질않습니다.
포스팅해주신 다음의 문장이요..
“단 친화크로마토그래피를 걸고 나서 SDS-PAGE 를 걸어보니까 잡스러운 단백질들이 많이 있어서 이걸 좀 더 정제해보겠어요~ 라고 그대로 그걸 이온교환크로마토그래피에 걸어버리면~ 아마도 대개의 경우 님의 목적단백질과 잡 단백질은 바로 컬럼에 붙지 않고 빠져버릴 것이다. 그러므로 이온교환크로마토그래피를 위해서는 일단 단백질이 들어있는 용액내의 염 농도를 낮추어야 한다. ”
어떤 이유때문에 컬럼에 붙지 않고 빠져나가는 것인지 쉽게 설명 부탁드릴게요..
긴 글 써주셔서 많은 도움이 됐습니다. 답변부탁드릴게요 감사합니다.
1. 물론 단백질에서 언제나 적용되는 원리는 ‘단백질은 그때그때 달라요’ 밖에 없습니다 (…) 즉 pH 변화에 민감하여 denaturation되는 단백질이 있을 수 있는가 하면, 어느정도의 pH의 변화에도 적어도 단시간적으로는 (실험을 할 수 있는 몇 시간 정도) 문제없는 단백질도 있습니다. 결국 자신이 정제하려고 하는 단백질의 성질에 맞추어 문제가 없으면 pH를 다소 낮추어 (어차피 ion exchange에 붙이냐 안 붙이냐 정도라면 pH 1-2 정도의 차이면 충분합니다) 정제를 시도해 볼 수 있습니다.
2. Ion Exchange에서 Elution이 되는 원리는 Counter-Ion, 즉 Anion exchange 라면 – charge를 띄고 있는 단백질보다 좀 더 강하게 Anion 컬럼에 붙는 -이온 (가령 NaCl solution이라면 Cl-) 과 단백질이 경쟁해서입니다. 그런데 보통 Ni-NTA라면 버퍼에 매우 높은 농도의 NaCl (최소 500mM 이상) 이 존재하죠. 당연히 이런 농도의 counter ion 이 존재할때 단백질은 ion exchange에 잘 붙지 않습니다. (counter ion이 더 잘 붙을테니까) 그래서 염 농도를 낮춰야 하는 것입니다.
ㅋㅋㅋㅋㅋㅋ 글 왜케 잘쓰세요