오늘날 현대인이 가장 널리 복용하는 약에는 ‘혈압약’, ‘고지혈증약’을 빼놓을 수 없습니다. 그만큼 자주 복용하지는 않을지도 모르지만 현대인의 일상에 필요한 약이 또 있죠. ‘발기부전치료제’ (…) 곧 비만치료제도 이러한 대열에 들어올지도 모릅니다.
너무 흔한 약이라는 점에서 때로는 이 약을 항상 복용하는 것이 그렇게 중요한가 하는 생각이 들기도 합니다만, 사실 이 약들은 인류가 만든 어떤 약물보다 인류의 수명을 연징한 약에 속합니다. 그리고 이러한 약들은 불과 100년 전, 아니 50년 전까지만 하더라도 치료가능한 질병이라고 여겨지지 않았던 질환들을 관리 가능한 질병으로 만든 핵심 요소가 됩니다.
그러나 그렇게 중요한 약들이 개발되기 위해서는 어떤 과정이 필요했는지, 그리고 어떤 연구과정이 필요했는지는 의외로 많은 대중들에게 잘 알려져 있지는 않은 듯 싶습니다.
이 책은 이러한 ‘지적 틈새’를 노린 책입니다. 어떻게 보면 글쓴이 당사자 (비만, 고혈압 환자;;) 의 입장에서 궁금했던 내용을 알아보고자 하는 일환이기도 합니다. 환자중심주의과학서
암튼 여러분이 먹는 고지혈증, 고혈압, 비만치료제 (그리고 발기부전치료제;;;) 의 과학에 대해서 알고 싶은 분은 읽어보셨으면 합니다
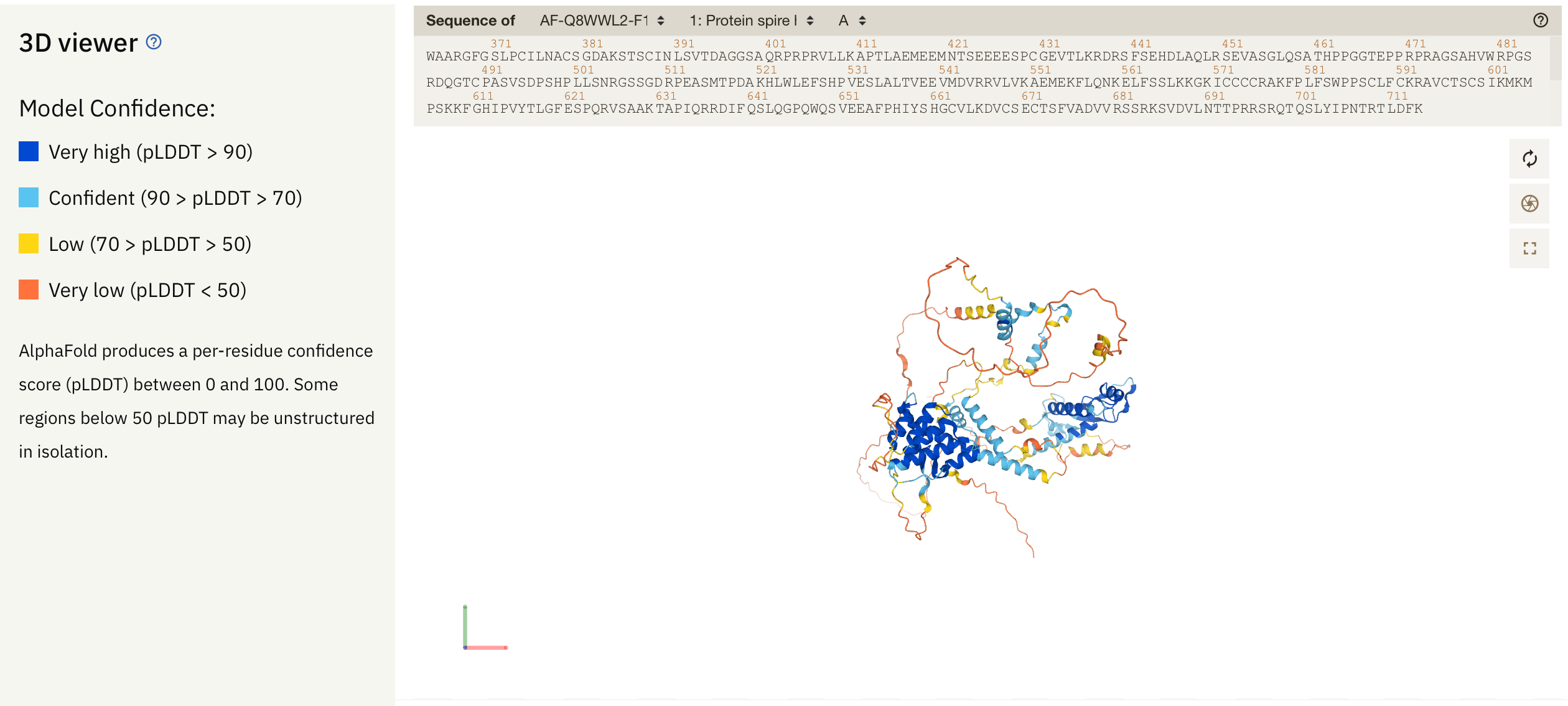
알파폴드 2 의 실행 가능한 코드와 예측 데이터베이스가 공개되면서 많은 사람들의 흥미를 끌고 있는데, 구조 예측의 정확성도 그렇지만 몇 가지 흥미있는 정보를 제공하고 있다. 즉 모델 예측의 신뢰도를 제공하고 있는데
per-residue confidence score (pLDDT) 라는 이름으로 단백질에서 모델 예측의 신뢰도를 제공하고 있는데, 90 이상은 매우 신뢰도가 높은 영역, 그리고 70에서 90은 어느정도 신뢰도가 있는 부분, 그리고 50-70은 신뢰도가 낮고, 50 이하는 거의 disordered 된 영역이라고 보면 된다. 그런데 이러한 pLDDT 데이터는 어디에 저장되어 있나.
PDB 파일을 받아서 이것을 텍스트 파일로 열어보면
다른 PDB 파일과 마찬가지로 원소의 종류, 아미노산의 종류, X,Y,Z 좌표 등등이 있고, 제일 오른쪽에서 두번째의 컬럼 (30.28, 24.69…) 에 바로 이 정보가 저장된다.
사실 이 컬럼은 B-factor 컬럼이라고 하여, temperature factor 라고 불리는 데이터를 저장하는 곳이다. 원래 이 부분은 실험적으로 결정된 단백질 구조에서 해당 부분의 원자의 위치가 평균적인 원자에 비해서 얼마나 벗어나 있는지를 기술하는 수치로써, 숫자가 높을수록 해당 부분의 구조가 Flexible 하고 불확실하다는 것을 의미한다.
그런데 많은 구조생물학자는 B-factor 컬럼을 다른 용도로도 많이 사용하는데, 특정한 단백질 부분이 여러가지 유사 서열 중에서 얼마나 보존되었는지의 수치를 계산하여 B-factor 컬럼에 넣기도 하고 (https://consurf.tau.ac.il) 여러가지 용도로 사용한다. 이와 마찬가지로 알파폴드의 예측에서는 pLDDT 값을 B-factor 컬럼에 넣어두고 있다.
그렇다면 이것을 이용하여 PyMol 등의 단백질 구조 비주얼라이즈 프로그램에서 어떻게 이용하는지를 알아보자. 우선 알파폴드 DB에서 예측한 데이터를 하나 다운로드해보자. (참고로 구조 페이지는 여기이다)
이것을 PyMol에서 불러보면
pLDDT 로 컬러링을 하려면 다음과 같이 B-factor에 기준하여 컬러를 칠하도록 설정한다.
오른쪽의 패널에서 개체에서 color/spectrum/b-factors를 선택하면
다음과 같이 B-factor 에 들어간 pLDDT 스코어에 따라서 색칠을 한다. 그런데 알파폴드 DB와는 달리 신뢰성이 높은 곳이 적색, 낮은 곳이 청색이므로 좀 헷갈린다. 이를 바꿔주려면, Pymol의 명령행에서
spectrum b, rainbow2_rev 라는 명령을 넣어주면
다음과 같이 알파폴드 2 DB에서 보는 것과 비슷하게 바뀐다.
만약 컬러 팔레트가 그리 마음에 들지 않는다면 여기에서 제공하는 여러 종류의 팔레트를 참고하여
혹은
너님 취향대로 색을 칠해주면 된다.
그런데 신뢰도가 없는 링커가 영 거슬린다. 화면에서 표시를 생략하거나 아예 날려버릴 수는 없을까? 이렇게 색을 칠한 다음에 서열에서 일일히 선택해서 감춰버릴수도 있지만 영 폼이 나지 않는다. 명령어 하나로 신뢰도가 낮은 영역을 선택해 버릴 수는 없을까?
select br. b<60
이 명령을 내리면 아미노산 residue 기준으로 b factor(실제로는 알파폴드가 넣어둔 pLDDT 스코어) 가 60 이하인 신뢰성이 없는 부분만을 선택한다.
이것을 숨겨버리면 간단하게 신뢰도가 낮은 부분을 선택하여 숨길 수 있다.
여기서 서열을 띄워놓고 추가적으로 선택을 하든가…
아니면 반대로 신뢰도가 높은 부분만을 골라서 선택해서 별도의 객체로 만들어 놓는 것도 가능하다. pLDDT 스코어 70인 부분을 선택한 후 이것을 객체로 만들어서 후속 분석에 사용…
혹은 객체만을 PDB로 export 하면 깨끗하게 잡스러운 영역이 사라진 구조가 나온다.
동시에 여러 개의 PDB 에서 신뢰성이 없는 부분을 필터링하고 싶으면 그냥 적절히 PDB 를 파싱하여 B factor 기준으로 Residue를 날려버리는 코드를 짜면 된다. 당장은 나는 그런 코드가 필요없으므로 안 짜겠지만 뭐 어려운 거 아니니 알아서 (…)
비록 출간에 코로나 바이러스 펜데믹 상황이 큰 역할을 한 것은 분명합니다만 본 책의 내용 중 상당수는 펜데믹이 발생한 2020년 초 이전에 바이오스펙테이터에서 ‘바이러스와의 전쟁’ 이라는 연재에서 다룬 내용입니다.
물론 코로나바이러스에 대한 내용은 그 이후에 쓴 내용이긴 합니다만..즉, 코로나바이러스 판데믹의 발생과 종식 여부와 관계없이, 인류는 그동안 바이러스와 지난한 전쟁을 치루어 왔으며, 이러한 싸움은 코로나바이러스 이후에도 계속될 것이다…가 책의 한 줄 요약이 되겠습니다.
여러가지 바이러스 질병이 있지만, 이 책에서는 20세기 이후 인명피해뿐만 아니라 사회적으로도 가장 큰 파급을 불러일으킨 3종의 바이러스, 즉 인플루엔자 바이러스, HIV, 코로나바이러스를 다룹니다. 어떻게 보면 바이러스 그 자체보다도 바이러스와 닝겐과의 싸움 연대기에 가까운 구성입니다. 즉, 바이러스라는 미지의 적과 만나서 이를 어떻게 식별하고 어떻게 극복하였는가? 에 대한 이야기가 되겠습니다.
그리고 인간의 지성이 발전하기 훨씬 이전부터 바이러스와 알아서 수억년부터 싸움을 벌여온 인간의 면역력에 대한 이야기도 어느정도 상세하게 다룹니다. (이를 위해서 HIV 에 대한 이야기를 2부로 다루었습니다)
사실 온 국민이 백신을 접종받는 상황에서 ‘전 국민이 바이러스와 면역의 전문가’ 가 된 것처럼 생각되는 경우가 있지만, 여전히 일반 대중의 면역에 대한 상식은 부족합니다. 그리고 면역에 대한 생각도 ‘항체의 유도’ 정도에 그치고 있습니다. 그러나 인간의 면역에서 ‘항체’ 는 극히 일부의 역할을 할 뿐이지만 ‘항체 이외의 면역’ 에 대해서는 대중의 이해가 상당히 부족하죠. 이러한 부분에 의해서도 어느 정도 기본적인 지식을 전달하려고 노력하였습니다. 아무튼 3부의 코로나바이러스에서 코로나바이러스에 관련된 많은 연구결과와 지식을 정리하려고 했습니다만, 이 책의 목적은 단지 코로나 바이러스 펜데믹 극복을 위한 교양의 전달이라기보다는, 이 참에 바이러스와 면역에 대한 교과서적인 상식을 어느정도 전달하려고 하였습니다.
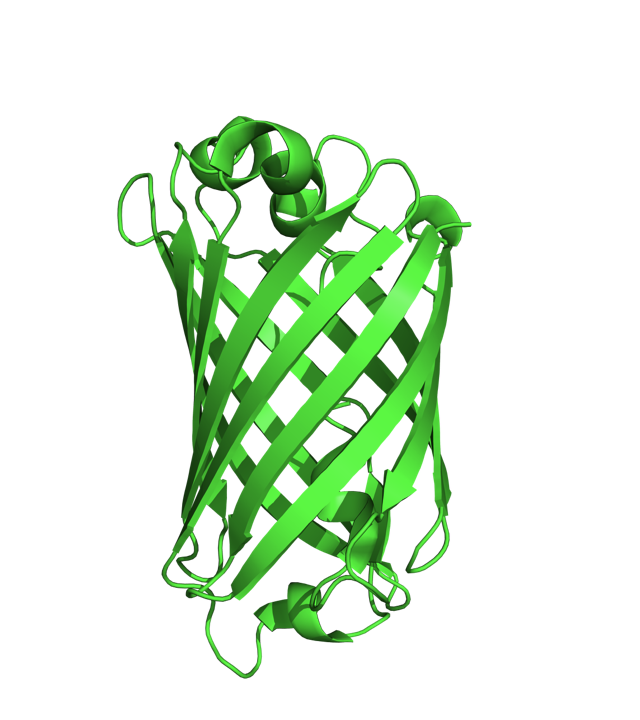
요즘은 단백질을 표시할 때 다음과 같은 리본 다이어그램 (Ribbon Diagram) 을 많이 이용한다.
즉 알파 나선(Alpha-Helix)을 리본 형태로, 베타 쉬트(Beta-Sheet) 를 화살표로, 그리고 루프를 연결선으로 그리는 이러한 스타일은 단백질의 2차 구조와 전체적인 형태를 간단하게 보여줄 수 있다는 점에서 많은 사람들이 좋아하고 있으며 수많은 문헌에서 사용되고 있다. 그러나 이러한 리본 스타일의 단백질 그림은 언제부터 나타났을까?
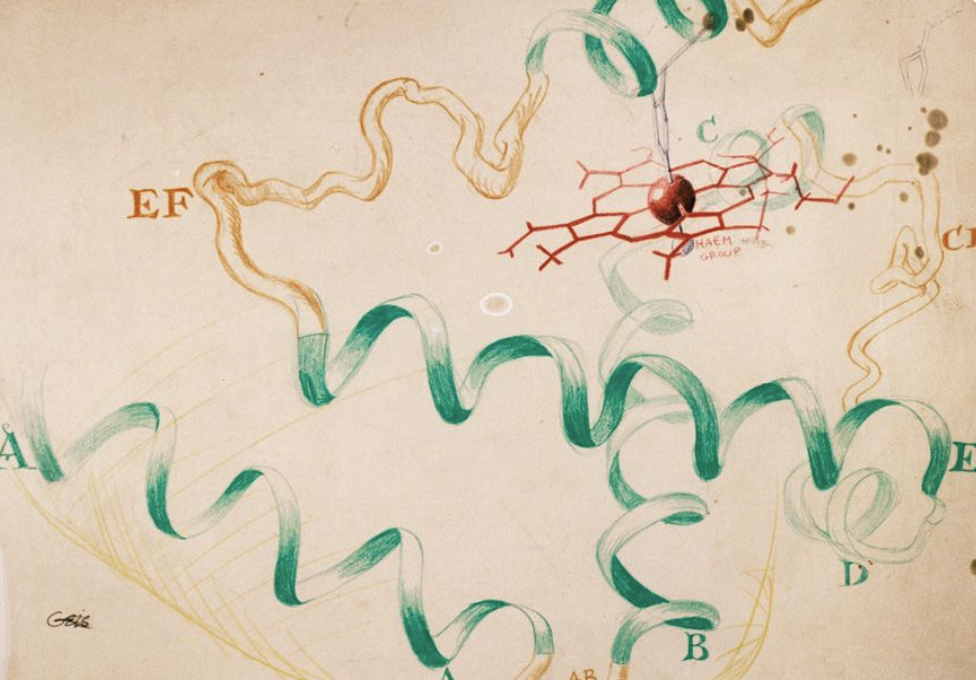
사실 최초의 단백질 입체 구조 규명인 1957년 존 켄드류의 마이오글로빈 구조 논문에서는 이러한 그림을 사용하지 않았다. 그는 마이오글로빈 결정에서 나타난 전자 밀도 데이터를 기반으로 다음과 같이 마이오글로빈의 구조를 묘사하였는데..
소….소세지?????
나중에 좀 더 고해상도의 데이터를 가지고 마이오글로빈의 원자 수준의 모델을 만들기 위하여 존 켄드류는 수수깡을 이용하여 다음과 같이 모델을 만들었고 화가 어빙 게이스 (Irving Geis)는 사이언티픽 아메리컨 잡지에 실을 목적으로 원자 수준의 다음과 같은 그림을 그렸다. (당시에는 컴퓨터 그래픽 따위가 없었으므로 ‘손’ 과 ‘붓’ 으로 그렸다)
이렇게 단백질을 구성하는 원자를 막대기로 그리는 방식의 표현은 단백질을 구성하는 모든 원자를 자세히 보여준다는 장점이 있었다. 그러나 이러한 방식의 ‘막대기’ 형식의 단백질 표현은 몇 가지의 문제가 있었는데
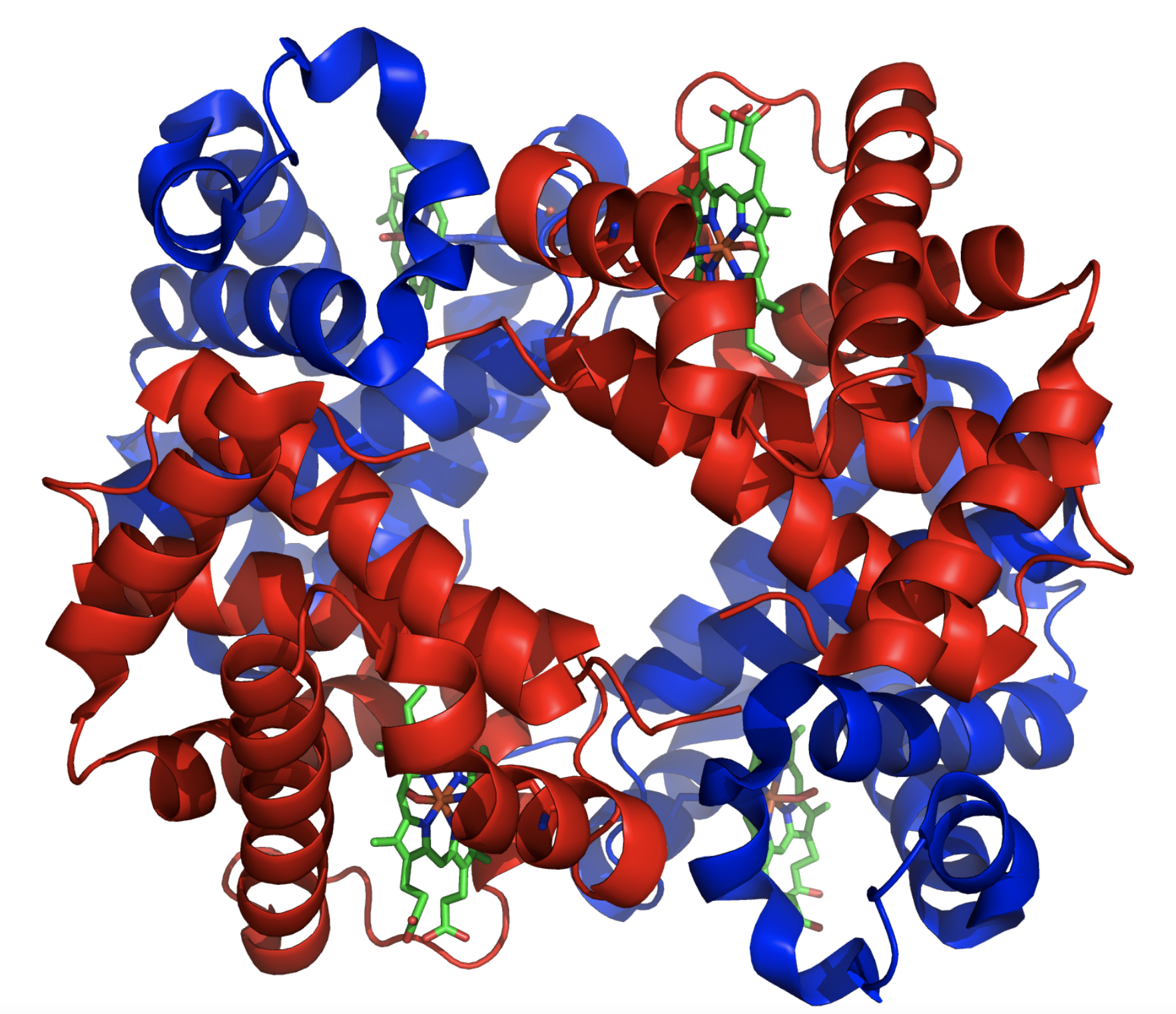
(1) 간단한 단백질이면 모르지만, 단백질이 복잡해질 수록 점점 전체적인 구조를 알기 힘들어진다. 가령 헤모글로빈만 하더라도 마이오글로빈과는 달리 4덩이의 단백질로 구성되어 있다. (2) 단백질의 2차 구조, 즉 알파 나선과 베타 쉬트가 어떻게 구성되어 있는지를 알기 어렵다.
사실 어빙 게이스는 최초로 단백질을 묘사하면서 알파 나선을 리본 형태로 묘사하는 것을 시도하였다. 가령 마이오글로빈의 구조를 묘사할 때 주로 알파 나선으로 구성된 단백질을 리본 형태로, 헴을 막대기로 묘사한 것을 처음 시도한 것은 어빙 게이스이다.
그러나 그는 단백질의 베타 쉬트를 화살표로 표시하지는 않았다. 그렇다면 알파 나선을 리본 형식으로, 베타 쉬트는 진행 방향을 화살표로 표시하는 현재의 ‘리본 다이어그램’ 을 완성한 사람은 누구인가?
지금 현재 듀크대학 생화학과에 약 50년 넘개 재직하고 있는 리처드슨 교수는 남편인 데이비드 리처드슨과 함께 1970년대부터 단백질 결정학을 연구하였다. 그가 1975년 최초로 규명한 과산화물 제거효소 (Superoxide Dismutase)는 다음과 같이 베타 쉬트가 많이 있는 단백질이었다.
리처드슨은 나중에 이러한 다이어그램을 손으로 그렸고, 이 그림은 단백질을 2차 구조로 간단히 표시할 수 있으며, 비슷한 구조를 가진 복수의 단백질의 전체적인 구조를 일목요연하게 볼 수 있게 됨으로써 매우 유용한 표시 수단이었다.
그러나 1970년대 말까지만 하더라도 이러한 리본 다이어그램은 손으로 그려야 하였으므로 그림에 재주가 있는 사람이 아니면 엄두를 낼 수 없을 상황이었다. 그렇다면 똥손도 이러한 그림을 그릴 수 있게 만들어준 컴퓨터 프로그램은 처음 어떻게 등장하였을까?
1982년 아서 레스크 (Arthur M Lesk)와 IBM의 칼 하드맨 (Karl D Hardman) 이라는 연구자는 단백질의 원자 좌표가 들어있는 PDB 파일을 입력 파일로 주면 이러한 다이어그램을 그려주는 프로그램을 개발하였다. 이것이 지금 현재 볼 수 있는 모든 단백질 입체 구조 시각화의 원조가 되는 프로그램인 셈이다.
지금의 기존으로는 상당히 조악한 그림이지만, 당시에 이러한 그림 그리기 위해서 화가에게 애걸복걸해서 겨우 몇 달 걸려서 한장 그리던 과학자들에게는 이렇게 데이터만 넣어주면 단백질 구조를 그려주는 컴퓨터 프로그램의 등장은 정말 획기적인 일이었을 것이다. 그 중요성에 걸맞게 이 논문은 1982년 S모 잡지에 출판되었다.
그 이후 수많은 단백질 시각화 소프트웨어들이 명멸하였다. Rasmol,Molscripts, MolMol, PyMol, Chimera, ChimeraX…지금은 웬만한 ㄸ컴에서도 리얼타임으로 돌아가는 단백질 시각화 소프트웨어지만 1990년대 중반만 하더라도 아무나 돌릴 수 없었고 그 당시에도 대당 수천만원이던 실리콘 그래픽스에서 나온 존나 비싼 워크스테이션에서나 그릴 수 있었다.
그러나 이러한 것이 지금과 같이 ㄸ컴에서도 다 돌아갈 수 있게 된 것은 과학자들의 불철주야에 걸친 노력….은 1도 관계 없고 1990년대 말부터 본격화된 3D 게임의 붐과 GPU등장으로 아무 컴퓨터에서나 3D 그래픽스와 리얼타임 그래픽을 즐길 수 있게 된 덕이다. 따라서 이를 가능케 한 영웅(?) 들에게도 인사를 드리는 것이 좋을 것이다.
생화학을 공부한 사람이라면 20종류(+2종) 의 단백질을 의미하는 3자 혹은 1자 코드에 대해서 알고 있을 것이다.
사실 3자 코드는 아미노산의 영어 이름의 첫 3글자로 거의 되어 있기 때문에 이것이 어디서 나왔는지 모르는 사람이 없다. 그러나 1자 코드는? 물론 세린 (Serine), 알라닌(Alanine), 류신 (Leucine), 메티오닌 (Methione) 등은 앞 글자로 되어 있기 때문에 외우기 어렵지 않지만 문제는 앞 글자와 다른 것들이다. 아르기닌 (Argnine) 이 R, 타이로신 (Tyrosine)이 Y 인 것은 이미 A와 T가 다른 아미노산이 사용하고 있기 때문에 두번째 글자를 사용했다고 하면 그래도 외워질 수 있으나 페닐알라닌은 왜 F이며 트립토판은 왜 W인가? 글루탐산은 왜 E 이며 아스파르트산은 왜 D 인가?
누가 어떤 의미를 가지고 어떤 목적으로 1글자 코드를 만들었을까?
를 궁금해 해 본 사람은 없는가?
일단 아미노산의 One-Letter Code를 제안한 사람은 오늘날에도 일부 학계에서 그 이름이 잘 알려진 분이다. 이 분은 바로..
이 분은 생물정보학에 조금이라도 관련이 있는 사람이라면 몰라서는 안되는 분인데, 그 이유는 거의 생물정보학의 창시자라고 불러도 될 분이기 때문이다. 아직도 ? 이라고 생각하는 사람이라면 치환행렬 (Substitution Matrix) 라는 개념을 생물정보학에서 처음 제창하였고 이 분이 만든 PAM (Point Accepted Mutation) 이라는 개념은 아직도 사용된다는 것 정도는 기억할 것이다. 이러한 개념을 처음 제안한 사람이 바로 이 분이다.
그렇다면 이 분은 어떤 배경을 가졌길래 생물정보학의 창시자라고 불릴 만한가? 자세한 문헌은 여기를 참고하도록 하고..
이분은 원래 양자화학 (Quantum Chemistry) 으로 1948년에 학위를 했는데, 연구 주제는 화합물의 공명에너지를 당시에 희귀했던 컴퓨터를 이용하여 계산을 하는 것이었다. 당시에 컴퓨터라는 것이 발명된지도 얼마 되지 않은 상황에서 컴퓨터를 잘 다룬다는 것이 그당시로는 얼마나 대단한 스킬이겠는가? 어쨌든 그는 박사학위를 마치고, 컴퓨터를 이용하여 화학 관련 계산을 하는 당시에는 흔치 않은 전문가가 되었고, 1960년 메릴렌드의 National Biomedical Research Foundation 이라는 곳에 자리를 잡았다. 여기서 컴퓨터 기술을 이용하여 여러가지 생물학적인 문제를 해결하는 다양한 연구를 하게 되었다.
그러던 중 그의 관심을 끌게 된 것은 ‘서열 정보’ 였다. 오늘날은 주로 DNA 서열을 먼저 결정하고 유전 암호에 의해서 번역된 서열로 단백질 서열을 유추하지만, 실용적인 DNA 서열 결정방법이 개발된 것은 1970년대 후반이고, 1960년대에 있던 단백질의 서열은 1952년 프레데릭 생어 (Frederic Sanger)가 개발한 단백질의 서열 결정 방법에 의해서 결정된 것이다.
물론 이 양반은 1977년에 DNA 서열을 결정하는 방법도 개발하긴 했지만, 여튼 1970년대 후반까지 인류가 알고 있던 단백질의 서열은 모두 정제된 단백질을 분해하여 어떤 폴리펩타이드로 구성되었는지를 알아내서 알아낸 것이었다. 1960년대 중반쯤 인간이 알고 있던 단백질 서열은 약 70종류였고, 이들은 대부분 사이토크롬 c 나 헤모글로빈과 같이 대량으로 많이 얻기 쉬운 단백질이었고, 여러 가지 다른 생물 유래의 것들이 많았다.
이들은 이렇게 얻어진 단백질 서열을 정리하면서 서로를 비교할 필요성을 느꼈다. 그렇다면 이것을 어떻게 비교해야 하는가? 기존에는 단백질의 아미노산 서열을 3자로 기록했었다. 그러나 당시의 후진 컴퓨터는 진짜로 보잘것 없는 기억용량을 가지고 있었으며, 기억용량을 줄이기 위해서는 아미노산의 글자를 1자로 하는 것이 절대적으로 유리했다. 그리고 여러가지 다른 생물의 서열을 비교할때 프린터로 출력하여 서열을 비교하는데도 3자 대신 1자로 비교하는 것이 보기 편했기 때문에 데이호프는 각각의 아미노산에 대응하는 코드를 만들기로 했다. 일반적으로 많이 사용되는 단백질의 아미노산은 20종이고, 알파벳은 26종이므로 적어도 아미노산에 대응하는 글자가 모자랄 염려는 없다.
일단 시스테인 (C), 히스티딘 (H), 이소류신 (I), 메티오닌 (M), 세린 (S), 발린 (V), 알라닌 (A), 글리신 (G), 류신 (L), 프롤린 (P), 쓰레오닌 (T)은 그대로 첫 글자를 사용하였다. 글자가 겹치는 아미노산들이 있는데 왜 이들을 선택했을까? 그 기준은 그때까지 알려진 단백질 서열에서 더 자주 사용되며, 구조가 간단한 아미노산에게 우선권을 주었다고 한다.
그러나 이제부터 첫글자가 겹치는 것이 등장하는데..
아르기닌 Arginine
타이로신 Tyrosine
이들은 두번째 글자인 R과 Y를 이용하여 지정하였다. 그러나 페닐알라닌 Phenylalanine과 트립토판 Tryptophan은? 두번째 글자인 R 역시 이미 사용되는 코드이므로 겹치지 않아야 한다. 페닐알라닌의 경우에는 비슷한 발음인 Fenyl…을 따서 F로, 트립토판은 화학구조에서 두 개의 링 (Double Ring)이 있다는 것에 착안하여 W(Double U, 더블유) 로 지었다.
그리고 다음은
아스파르트산 Aspartic Acid
아스파라긴 Asparagine
글루탐산 Glutamic Acid
글루타민 Glutamine
아미노산의 화학 구조를 아는 사람이라면 아스파르트산과 글루탐산에 암모니아가 하나 더 붙으면 아스파라긴과 글루타민이 된다는 것을 알고 있다. 따라서 아스파르트산과 아스파라긴, 글루탐산과 글루타민은 뭔가 연관이 있는 것이 좋다. 그리고 아스파르트산은 글루탐산보다 작고, 아스파라긴은 글루타민보다 작다는 것을 떠올렸다.
따라서 남은 글자 중에서 아스파르트산을 D, 글루탐산을 그 다음 글자인 E로 정하였다. 그리고 분자량이 더 큰 (아미노기가 붙어서) 아스파라긴과 글루타민을 같은 순서로 남는 글자인 N과 Q로 정하였다고 한다. 왜 N 다음의 글자가 아닌 O가 아닌 Q를 글루타민으로 정했을까? 정확한 이유는 알 수 없지만 Q의 발음이 Glu와 그나마 유사해서 그런 것일지도 모른다. (O는 한참 나중에 22번째 아미노산인 파이로라이신의 약자로 사용된다)
마지막으로 남은 것은 라이신 Lysine 으로써 크기가 비슷한 류신 (Leucine,L) 에서 그리 멀지 않은 곳에 위치한 K를 선택하였다.
U를 사용하지 않은 이유는 손으로 쓴 글자를 해독할때 V와 혼동되지 않도록, O는 프린터 인쇄가 잘못되는 경우 G,Q,C,D와 혼동될 수가 있어서였다고 한다 (지금은 상상하기 힘들지만 그때는 빈번히 아미노산 서열을 손으로 쓰거나 도트 프린터로 인쇄하던 시대라는 것을 생각해야 한다. 결국 이 두 가지 코드는 그런 문제가 없어졌을때 셀레노시스테인과 파이로라이신의 코드로 채용된다)
그런데 사실 아미노산을 1자로 표기할 생각을 한 것이 아니었다. 적어도 수 명의 학자가 자기만의 1자 코드를 주장했었는데, 1963년의 키모트립신의 구조를 다룬 논문에서는 우리가 지금 사용하고 있는 1자 코드와 다른 코드를 사용하고 있다는 것을 볼 수 있다.
페닐알라닌에 F를 사용한 것은 같지만 류신에 U, 이소류신에 W, 아스파르트산에 A, 글루탐산에 G…등 오늘날 알려진 코드와 공통적인 것이 별로 없다. 그러나 아스파라진과 글루타민에 N과 Q를 사용했고 아르기닌에 R을 사용했다는 것은 오늘날의 코드와 동일하다. Dayhoff와 Eck이 자신의 코드를 만들 때 여기에 얼마나 영향을 받았는지는 모르겠지만, 몇 가지 아이디어를 빌려왔을 가능성은 있다.
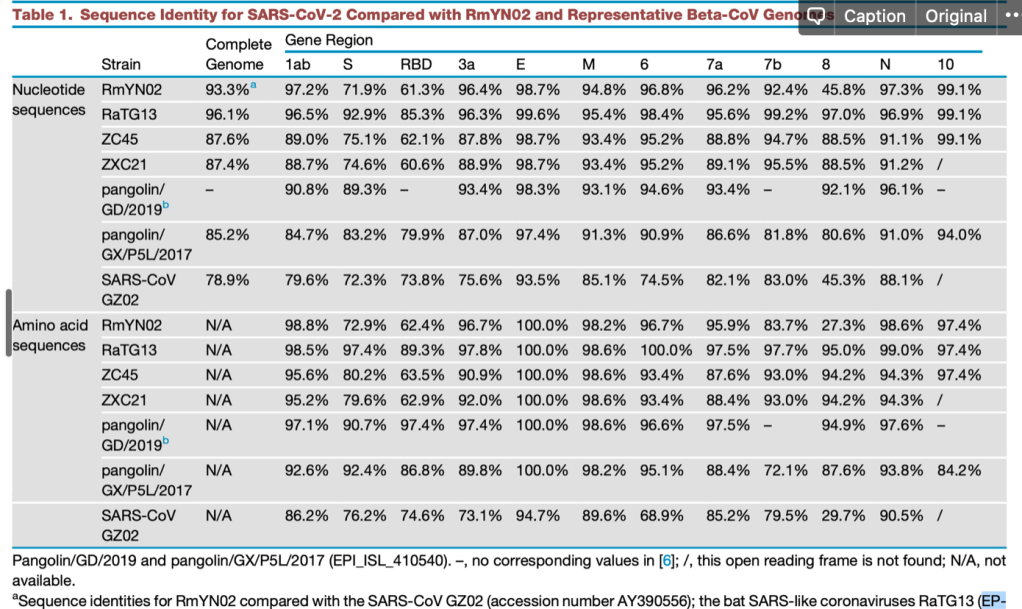
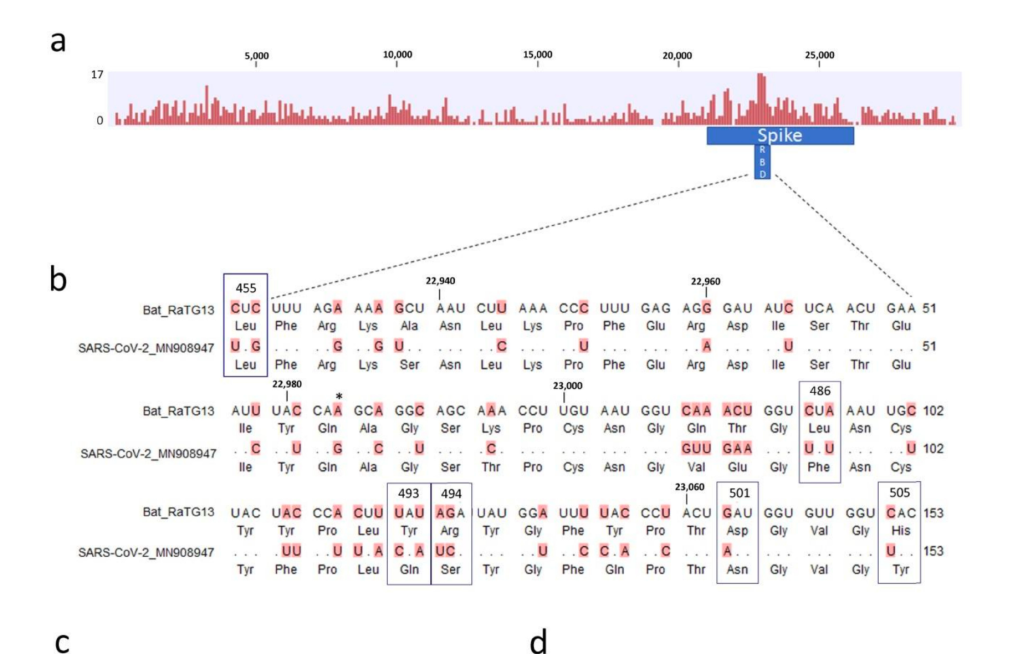
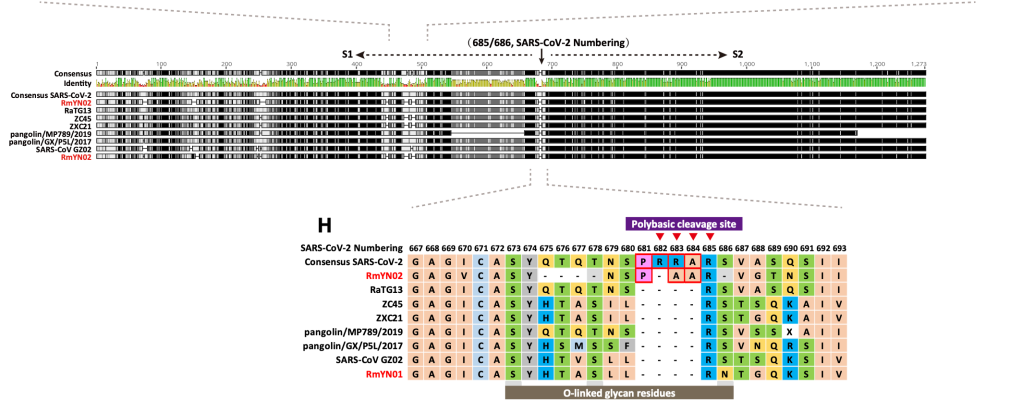
현재까지 SARS-CoV-2와 가장 가까운 바이러스인 RaTG13 은 전체 지놈 기준으로 핵산 서열의 일치도가 96.1% 입니다. 그 이야기는 3.9% 의 핵산 서열이 다르다는 것이고, SARS-CoV-2 의 지놈이 길이가 30,000 염기이므로 약 1,000염기 이상이 다르다는 것입니다. 이 중 가장 상동성이 떨어지는 것은 바이러스의 스파이크 단백질 유전자인 ‘S’ 유전자이고, 그 중에서도 직접 세포의 수용체인 ACE2 와 결합하는 영역인 RBD 부분입니다. 이 부분이 왜 가장 빈번하게 바뀌는가? 는 쉽게 유추할 수 있습니다. 많은 종류의 숙주에서 증식하려면 일단 세포에 침투해야 하고, 이 부분과 숙주 세포의 수용체와의 상호작용이 필수적입니다. 그런데 숙주의 수용체는 다 다르고..이를 위해서는 여기에 돌연변이가 많이 일어나서 각각의 환경에서 최적의 환경을 보이는 넘들이 선발된 것입니다.
당연한 것입니다만, 많은 핵산 변이가 다 아미노산 변화를 유발하지는 않습니다. 돌연변이가 유발되어도 아미노산의 변화를 유발하지 않는 synonymous mutation (코돈의 세번째에 주로 생기는) 가 아미노산을 바꾸는 변이보다도 더 많습니다. 그 이유는 어쩌면 당연합니다. 돌연변이가 생기면 바이러스의 생존 자체에 문제가 있는 부분과, 바뀌어도 좋은 부분이 있습니다.그나마 많은 돌연변이가 생기는 RBD 부분에서도 아미노산에 돌연변이가 생기는 것은 한정적입니다.
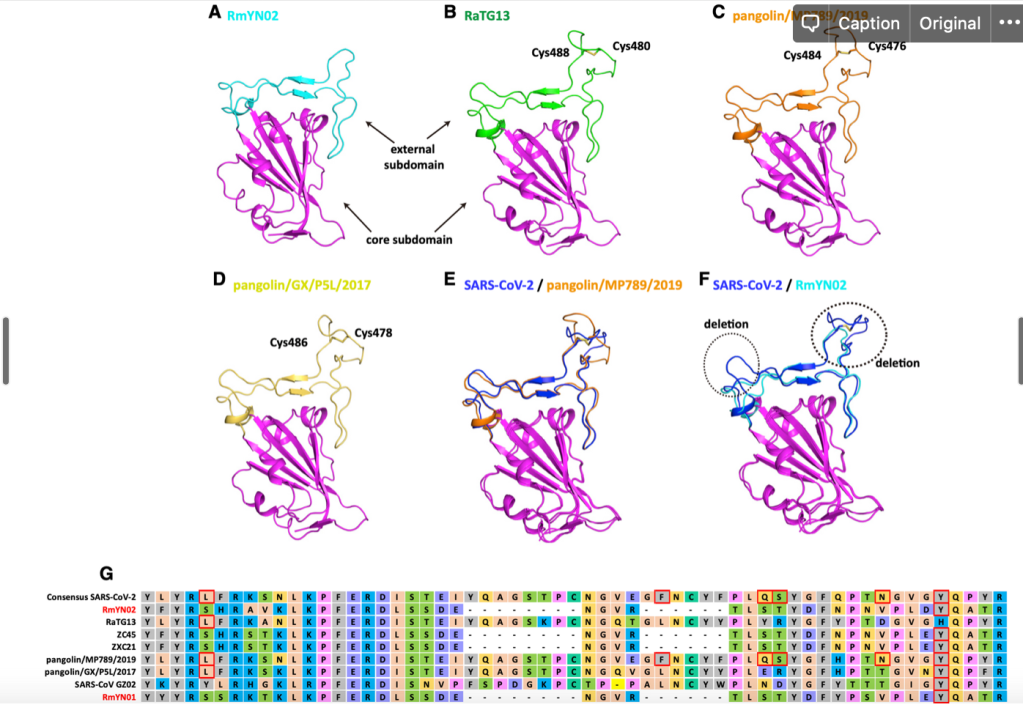
RBD 도메인에서 일어나는 변화는 단순히 아미노산 바이러스 변화뿐만 아니라 삽입된 서열 (혹은 삭제된 서열) 이 있습니다. 박쥐에서 발견된 일부 바이러스에는 SARS-CoV-2 나 RaTG13 에는 존재하는 삽입된 서열이 존재하지 않습니다.
이러한 서열의 삽입과 추가는 바이러스마다 다 ‘그때 그때 달라요’ 입니다. 가령 RMYN02 라는 바이러스는 RBD 도메인의 삽입 서열이 없다는 것에서 SARS-CoV-2 와는 다르지만, 스파이크 단백질의 다른 부분에서 SARS-CoV-2 에만 공통적으로 있다고 생각하던 삽입 서열이 있습니다.
위에 보면 SARS-CoV-2 에는 PRRA 서열이 있고, 일부는 이것이 혹시 인위적으로 조작된 증거가 아닌가 하는 궁예를 하기도 했습니다만, 뭐 자연계에 있는 다른 바이러스에도 이 부분에 삽입되는 경우가 있네요.
어쨌든 SARS-CoV-2 와 가장 가까운 자연계에서 발견된 바이러스와 SARS-CoV-2 에는 약 4% 의 서열 차이가 있고, 이는 1,000 염기 정도의 차이입니다. 염기의 차이 중 거의 대부분은 아미노산을 바꾸지 않는 돌연변이이고, 일부만 아미노산을 바꿉니다. 이들 아미노산 변이 중에서 어떤 것이 해당 바이러스들이 가지는 차이 (가 있다면) 에 기인하는지는 잘 모릅니다.
이제 이런 상황을 생각해 보도록 합시다. 어떤 매드사이언티스트 (…)가 자연계에 있는 코로나바이러스를 분리하여 이것을 뜯어고쳐서 닝겐에게 더 잘 퍼지는 바이러스를 만들어서 생물학 테러 같은 것을 일으키고 싶다고 해 봅시다. (왜? 매드사이언티스에게 이유를 묻지 마십시오. 매드사이언티스트는 특별히 자신에게 이익이 없어도 그런 헛짓거리를 하는 닝겐들입니다. 그래서 ‘매드’ 죠) 일단 바이러스 유전체를 인공적으로 조작할 수 있는 기술 자체는 있습니다.
즉 바이러스를 DNA 형태로 만들어서 플라스미드 벡터에 넣고, 시험관에서 Transcription을 하여 RNA를 만들고 감염시키는 것입니다. 이러한 것은 바이러스를 연구하는 분들이 많이 사용합니다.
자, 이렇게 자연계의 바이러스에 돌연변이를 주고 조작을 할 준비를 했다고 합시다. 어디를 뜯어고치겠습니까? 감염력을 증대시킨다면 스파이크 단백질, 특히 RBD 도메인 등에 인위적인 조작을 주고, 조작된 바이러스를 만들어서 감염력을 테스트해서 제일 잘 감염되거나 병리학적으로 독성이 제일 강한 넘을 선발할 수는 있겠지요. 그렇다고 한다면 스파이크 단백질이나 RBD 도메인에 돌연변이 라든지 인위적인 조작의 흔적이 남겠지요. 특별히 돌연변이를 준다고 해도 결국은 아미노산을 바꾸는 돌연변이여야 의미가 있을 것이므로 아미노산이 바뀌지도 않는 돌연변이를 만들지는 않을 것입니다. 바이러스의 다른 부분을 뜯어고친다? 아이고….스파이크 단백질 뜯어고치기도 힘든데 다른 부분은 또 왜…뭐 일단 스파이크 단백질을 뜯어고치고 다른 부분을 더 ‘개량’ 한다고 하더라도 그때 역시 아미노산이 바뀌지도 않은 돌연변이를 넣지는 않을 것이고 극히 한정적으로 아미노산이 바뀌어서 바이러스의 성질이 바뀌는 그런 것을 찾을 것입니다. 엔지니어링을 할 때 굳이 ‘잘 돌아가는 다른 부분’ 을 고칠 필요는 없으니까요. 어차피 이런 부분들은 여러 코로나바이러스에서도 스파이크 단백질에 비해서 많이 바뀌지도 않았습니다.
그렇게 만들어진 바이러스는 아마 스파이크 단백질의 일부분, 혹은 지놈의 다른 부분에서 극히 일부분만 한정적인 돌연변이가 있는 그런 바이러스가 될 것입니다. SARS-CoV-2 와 RaTG13 의 차이처럼 약 천개 넘는 돌연변이가 있고, 대부분은 기능에 영향을 미치지도 않는 아미노산을 변화시키지도 않는 돌연변이가 있는..
물론 바이러스를 ‘조작’ 하는 방법은 현대 유전공학적인 방법만 있는 것은 아닙니다. 사실 닝겐은 현대 유전공학이 대두되기 전부터 바이러스를 ‘조작’ 해 왔습니다. 무슨 말이냐구요? 약독화 백신입니다. 즉, 상당수의 백신은 살아있는 바이러스입니다. 이를 만드는 방법은 바이러스를 자신이 병을 일으키는 숙주와는 조금 다른 환경에서 존나 오래 키우는 것입니다. 바이러스를 오래 키우다 보면 돌연변이가 누적되고 그 성질이 변합니다. 즉 병을 일으키는 숙주와다른 환경에 살다 보면 자연스럽게 여기에 적응하는데 유리한 돌연변이가 축적되고, 이러한 돌연변이는 원래의 병원성을 약화시킬수도 있습니다. 이런 식으로 만들어진 백신 중의 대표적인 것이 황열병 백신으로써, 황열병 백신은 원래의 숙주인 원숭이 혹은 닝겐이 아닌 수정란에서 오랫동안 계대배양을 하여 병원성을 잃어버리도록 한 바이러스입니다. 또 다른 것은 소아마비 백신 중 경구 투여를 할 수 있는 생백신으로써 이것은 사빈이라는 사람이 만들었습니다. 이 역시 발견된 소아마비 바이러스를 존나 오랫동안 배양해서 얻어졌습니다.
그렇다면 병을 일으키는 바이러스와 인공적으로 존나 오래 배양되어서 얻어진 생백신은 얼마나 차이가 있을까요?
2002년의 이 논문에서는 존나 쎈 소아마비를 일으키는 바이러스인 마호니 스트레인 (Mahoney Strain)을 오랫동안 계대배양해서 만들어진 약독화 바이러스 (생백신) 의 염기서열 차이를 분석했습니다. 마호니 스트레인과 약독화 바이러스는 핵산 기준으로는 54개가 다르고 20개의 아미노산이 바뀌었는데, 약독화에 관여하는 아미노산 변화는 딱 2개였습니다. 사빈은 이 바이러스를 얻기 위하여 많은 사람들의 쿠사리 (너님은 언제 백신 만들래? 소크라는 사람은 벌써 다른 방법으로 만들었구만) 를 먹으며 몇년동안 존버하면서 바이러스를 배양 또 배양해서 이걸 만들었습니다. 소아마비 바이러스의 경우 지놈 사이즈가 7000 염기 정도입니다.
만약 이런 식으로 존나 바이러스를 배양해서 더 짱센 넘을 선발하는 식으로 자연적으로 일어나는 돌연변이에 의존해서 바이러스를 찾으면 얼마 정도 걸릴까요? 소아마비 바이러스와 코로나바이러스는 아마 돌연변이율이 다를 것이기 때문에 직접적인 비교를 하기는 어렵습니다. 그렇지만 뭐 이 글이 진지빨면서 하는 것은 아니므로 (…) 소아마비 바이러스와 코로나바이러스가 비슷한 속도로 돌연변이를 일으킨다고치고, RaTG13 에서 SARS-CoV-2 의 변이만큼을 나올 시간은…아마 사빈이 저걸 얻을 시간의 20배 (단순히 돌연변이 갯수의 비율. 다를수도 있음) 정도는 걸릴지도 모르겠군요.
그런데 우리는 이미 많은 양의 SARS-CoV-2 의 지놈을 시퀀싱하였고, 이를 통하여 사람을 통해서 퍼질 때 이 바이러스가 어느 정도의 속도로 변화하는지 짐작가능합니다. 지금 코로나바이러스가 수천만명의 사람을 통해서 퍼지고 있고, 이러한 상황에서 코로나바이러스는 대충 1년에 20염기 정도로 변한다고 합니다. 그렇다면 이런 속도로 ‘사람 중에서’ 퍼질 때 약 천개 정도의 돌연변이가 생기려면…50년?
그런데 이 상황은 수천만명의 실험대상(?)을 통해서 바이러스가 퍼졌을때 얻을 수 있는 상황입니다. 만약 매드사이언티스트가 연구실에서 한정적인 실험동물, 혹은 배양세포를 통해서 행한다면 어떻게 될까요. 물론 바이러스가 배양세포에서의 증식은 닝겐에서 자라는 것보다는 더 빠를 것이므로 증식이 빠르면 돌연변이 속도도 빨라질 수는 있습니다. 그러나 일단 존나 큰 스케일로 실험을 해야하겠지요. 지놈의 4% 를 바꾸는 것은 쉽지 않습니다.
좀 더 빠른 속도로 돌연변이를 얻기 위하여 돌연변이 유도를 하는 화합물이나 X선 같은 것을 써 볼까요. 물론 그렇게 하여 돌연변이 속도를 높일 수는 있을 것입니다. 그러나 그것이 더 빠른 표현형의 변화를 얻는 좋은 방법은 아닐수도 있습니다. 일단은 그렇게 얻어진 돌연변이 바이러스가 실제 표적이 되는 곳에서 어떻게 활동할지는 ‘실험’ 을 해봐야 압니다. 그걸 어떻게? 얻어진 돌연변이 바이러스를 막 주변에 퍼트려서 전파력을 검사한다? 현실적인 방법은 아닙니다. 세포 시스템에서 바이러스의 감염력을 조사하는 것도 대안입니다만, 암튼 수년 동안 정량적으로 조사해야겠지요.
어쨌든 유전공학적인 방법을 사용하든, 존나 오래 배양하면서 돌연변이를 기대하든 이 정도의 유전적인 변화를 닝겐이 일으키는 것은 쉽지 않습니다. 닝겐이 동물이나 식물을 육종할 때는 서로 교배하면서 유전체를 쉐킷쉐킷- 해주는 그런 꼼수가 있었고, 실제로 이렇게 교배를 통하여 유전 정보를 교환하는 것은 유전정보의 다변화에 좋은 방법입니다. 그러나 바이러스라는 쉑히들은 찐따라서 유전자 교환의 주요 방법인 쒝스도 못합니다….쒝스도 못하는 찐따새끼들….혹시 다른 유전정보를 가진 바이러스들이 동시에 감염되면 복제되는 와중에 유전정보 교환이 일어나지 않을까요? 실제로 바이러스의 지놈들을 찾아보면 서로 다른 종간의 바이러스 간의 재조합의 흔적이 보이기는 합니다. 그러나 그리 일반적으로 일어나는 일은 아닐 것입니다. 모르죠…세포주를 조작하여 바이러스 간 유전정보 교환,즉 쒝스를 위해서 또 다른 시스템을 설계….아 너무 복잡합니다.
이러다 보면 아무리 대단한 매드사이언티스트라도 현타가 몰려올 것입니다. "여기는 누구인가 나는 누군가 나는 뭘 하는가" 사빈 아재야 백신을 만들기 위한 목적이라도 있었지만 RaTG13 에서 SARS-CoV-2 으로 바이러스를 바꾸는 것은 도대체 어떤 목적인지도 모릅니다.
그나마 바이러스 유출의 시나리오로 가장 그럴싸한 것은 SARS-CoV-2 가 이전에 동물 등에서 분리된 (박쥐 등이 코로나바이러스의 소스였으므로 박쥐 등에서 바이러스를 수집하는 연구는 많이 진행중입니다) 기 분리된 바이러스였는데 연구소에서의 어떤 문제 (실수? 등등) 로 바이러스가 유출되는 정도일 것입니다. 물론 이것을 입증할 자료는당연히 없습니다. 그러나 어쨌든 인위적인 바이러스 개조보다는 훨씬 더 쉬워보입니다.
어쨌든 요약을 하면 이렇습니다.
현재까지 알려진 바이러스로부터 SARS-CoV-2 를 인공적으로 만드는 것은 존나 어렵고 사실 그럴 목적도 모르겠다.
존나 오래 인공적으로 배양하면 변화된 바이러스를 얻을수 있을지는 모르겠지만 그렇게 하려도 최소 수십년은 걸릴 것이다.
차라리 이전에 자연계에서 분리된 바이러스가 유출되었을 가능성이 1,2보다는 높다. 물론 3을 입증하는 증거는 아직까지는 없다.
바이러스를 개조해서 우리가 원하는 능력을 만들게 하는 것은 어렵다. 생명공학을 과대평가하시는 것은 업계인으로 감사합니다만 우리는 아직 그정도의 능력이 없는 찐따들입니다 ㅠㅠㅠ
“해당 분야 전문가가 보기에는 내용이 빈약한데, 일반인이 읽기에는 어렵지 않은가?”
“책의 독자를 어느정도로 상정하고 책을 쓰는지 모르겠다”
굳이 말하자면 내가 쓰는 책은 이 블로그 ,혹은 페이스북 페이지를 즐겨 찾는 독자 정도를 대상으로 쓰고 있다. 그렇다면 이 블로그 (페이지) 를 찾는 독자층은 어떨까? 실제로 이런 것을 조사해 본 적은 없다 (…) 그러나 대략적으로 덧글을 다는 분들은 현업 연구자, 대학원생, 이공계 대학생 등이 많이 계신 듯 하며 혹은 과학 공학 관련 업계 종사자들도 꽤 계신다. 과학에 관심있는 일반인들도 꽤 있을 것으로 생각하나 그 비율은 잘 모르겠다.
대략적으로 그 정도의 독자가 읽겠거니 하고 생각하고 글을 쓰는 셈이다. 한마디로 전혀 과학을 모르는 사람을 대상으로 쓰는 글과 책은 아니라는 이야기이다.
기본적으로는 대학 1학년 정도의 일반생물학 (혹은 고등학교 생명과학 2?) 정도의 지식을 가지고 있다고 전제하고 글을 쓴다고 생각하면 될 것 같다. 어떻게 보면 이 정도의 과학 지식을 갖추고 있지 않은 많은 사람들에게는 어렵다고 느껴져도 할 말이 없다. 그리고 대학 1학년, 고등학생 이하의 지식을 갖춘 사람들을 위해 쉽게 과학 지식을 설명하는 책 혹은 블로그도 필요할 것이고 중요할 것이다.
그러나 중요한 것은 그런 일은 내가 주로 할 일은 아니라는 것이다. 이미 그런 레벨에서 책을 쓰시는 분은 많지 않은가?
가령 가장 최근에 나온 ‘세포’ 는 모 대학교에서 개설된 ‘현대생물학사’ 라는 강의 준비를 하면서 썼고, 그 강의의 내용이 상당수 나온다. 해당 과목은 내가 처음 개설한 과목이 아니므로 어떤 의도로 개설되었는지는 잘 모르겠고, 명백히 과학사학자가 아닌 내가 이런 이름을 가진 과목을 강의하는게 과연 최선인가 하는 생각은 들지만, 여튼 이 과목에서는 나는 일반생물학 수준의 생명과학 교과서에 나온 결과들이 어떤 과정을 통하여 발견되었냐 하는 약간의 연대기적 맥락을 소개한다. 그런 것을 굳이 소개하는 이유는, 과학 전공에 입문하는 학생들이 단순한 사실의 암기를 넘어서, 교과서에 기술된 발견들이 어떤 과정을 통하여 발견되었는지를 약간이라도 이해한다면, 배우는 과목을 단순한 지식의 암기와는 조금 다르게 생각할 수 있지 않을 까 하는 기대 때문이다. 그리고 과학에 흥미를 느끼고 머학원에 샥..아님
참고로 이 강의의 수강생은 1학년 학생이 주이긴 하지만 2,3,4학년 학생도 있고, 주로 ‘생명과학부’ 에 다니는 학생들이다. 즉 기본적으로 일반생물학 정도는 수강한 학생들이다. 이 학생들을 기준으로 한 강의이고 여기에 기반한 내용이기 때문에 당연히 이러한 지식이 부족한 독자라면 어렵게 느껴질 수도 있고, 반대로 책에서 다루는 분야를 직접 연구하는 연구자라면 내용이 부족하게 느껴질 수도 있다 (애초에 이러한 연구자라면 자신의 연구분야를 개관하는 내용을 대중서에서 기대하는 것 자체가 조금 무리 아닐까. 리뷰를 보세요;;) 그러나 그 ‘애매함’ 은 어쩌면 의도된 애매함일 것이다.
즉 내가 쓰는 책은 여태까지도 그렇고 앞으로도 그 ‘의도한 애매한 범위’, 즉 일반인은 조금 어렵게 느껴질수도 있지만 해당 분야의 전공자라면 당연하게 느껴질 그런 범위에 머물고자 한다. (어차피 어려운 대중서를 아무리 읽어도 일반인이 전공자가 될 수는 없을 것이다. 그렇다고 완전한 전공자를 위한 책은 현실적으로 국내에서 출판을 하기가 쉽지는 않을 것이고)